Huntington’s Disease Fact Sheet
CIRM funds many projects seeking to better understand Huntington’s disease and to translate those discoveries into new therapies.
Description
In the U.S. about 30,000 people have been diagnosed with Huntington’s Disease (HD) and another 150,000 have a 50 percent risk of developing the disease because they have one parent who has or had HD. There are no effective therapies and the disease is uniformly fatal, usually in 10 to 20 years.
Huntington’s Disease results from a mutation to one gene, but that mutation can vary between patients and that may be linked to differences in how the disease progresses in various patients. But in all cases the mutated gene produces a protein that is toxic to nerve cells and eventually kills them. California’s stem cell agency has funded several projects that probe into both the nature of the mutation and ways to prevent or repair the damage from the mutant protein (a full list is below).
One obstacle to finding and testing potential therapies for HD is the lack of a good laboratory model that captures the complexity of the disease in people. So researchers are using stem cells to create a “disease in a dish” model, that allows them to create cells that reflect different forms of Huntington’s disease and then use them to screen different drugs to see if they are effective against HD. These teams are taking advantage of adult cell reprogramming to create so called induced Pluripotent Stem Cells (iPS) from patients with HD to see how the cells differ. One team, over time, also hopes to genetically modify these iPS cells so that they produce the correct Huntington protein and could be used as a personalized therapy, custom-designed for each person with HD, to reduce the chance their own immune system will destroy the new cells.
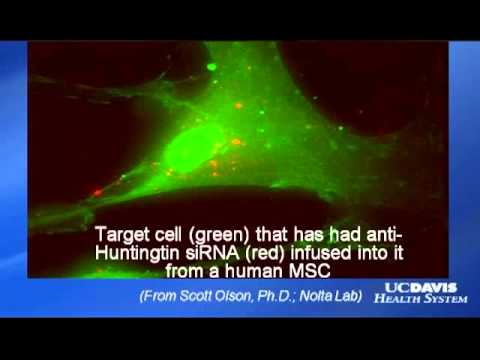
Two other CIRM-funded teams are seeking to identify potential therapies that could be delivered to the damaged nerves with stem cells. One proposes to use embryonic stem cells, mature them into early forms of those nerve cells and genetically modify them to deliver a compound that could protect patient’s other nerves from the toxic protein. The other plans to use a type of stem cell found in bone marrow, mesenchymal stem cells, to deliver a genetic fragment called iRNA to the nerves and shut down the faulty gene.
Clinical Stage Programs
University of California, Davis
This research team plans to use bone marrow derived mesenchymal stem cells to deliver a growth factor to patients’ damaged and endangered nerves. The factor they have chosen, called BDNF, has been shown to be effective in laboratory studies in reducing nerve cell death and improving the function of nerves. They completed an observational phase trial to monitor disease progression in a group of patients and have decided to conduct additional laboratory work with their cell-plus-gene candidate therapy.
CIRM Grants Targeting Huntington’s Disease
CIRM Huntington’s Disease Videos

















News and Information
- Stem Cells Show Promise For Treating Huntington’s Disease (ScienceDaily)
- UC Davis researchers awarded new stem cell grants (UC Davis)
- Race against the clock (UC Irvine)
- Overview of stem cell research for Huntington’s Disease (HDSA)
Resources
- NIH Huntington’s Disease Information
- Find a clinical trial near you: NIH Clinical Trials database
- Huntington’s Disease Society of America
- Family Caregiver Alliance
- National Family Caregivers Association
- The Movement Disorders Society
Find Out More:
Stem Cell FAQ | Stem Cell Videos | What We Fund